Amyloidosis or Beta-Fibrillosis
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
AMYLOIDOSIS or BETA-FIBRILLOSIS
AMYLOIDOSIS or BETA-FIBRILLOSIS
· Extracellular deposition of a peculiar hyaline, eosinophilic, Lugol- and PAS- positive substance, referred to as "amyloid" by Virchow.
· Staining by Congo red: red with green birefringence by polarized light interaction
· Rubbery consistency, fatty-like appearance, initial perivascular localization
· Poorly antigenic
· Localized or systemic deposits. They may cause atrophy of the parenchyma and dysfunction of affected organs.
· Over 20 types of amyloidosis have been reported, on the basis of different proteins forming amyloid fibrils
· Systemic forms mainly affect kidney-cardiac muscle-liver-spleen- accompanied by an increase of the organ size
· Composition and structure: proteins composed of heterogeneous amino acidic composition. The beta-pleated plane structure (X-ray diffraction) form fibrils which bind to acid mucopolysaccharides
Amyloid composition
1. Fibrillar protein component
· Constant beta-sheet fibrillar conformation: two spirally wound polypeptide chains held together yielding folded sheets
· Variable biochemical composition
· Often deriving from precursors of proteins with physiological roles
· Poorly immunogenic
· Protease resistant
2. Globular plasma protein component
· Globular glycoprotein (serum amyloid P) from the C-reactive protein
3. Glucosaminoglycans
· Create bonds between protein components
· The typical basal membrane heparan sulfate is frequently encountered
Artery section from an Amyloidosis-affected patient stained by Congo Red
Analyzed using A - normal light
B - polarized light
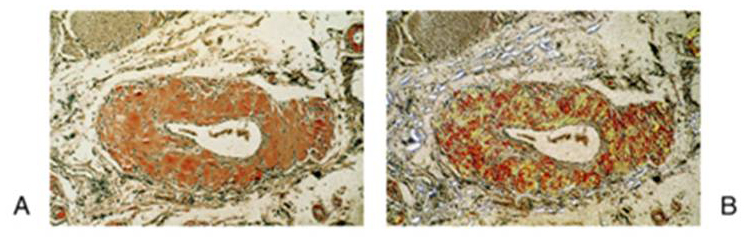
Rubin's Pathology Copyright 2006
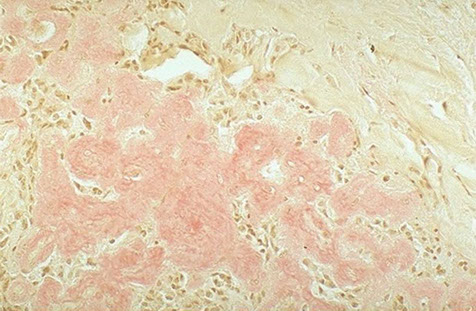
Light microscopy
Hyaline appearance of Congo red-stained amyloid deposits
ELECTRON MICROSCOPY IMAGE OF AMYLOID ACCUMULATION
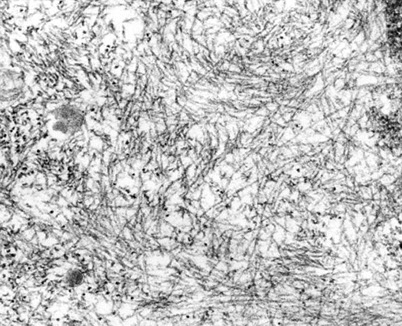
Rubin's Pathology- Copyright 2006
X diffraction
β-pleated structure of amyloid fibrils: the polypeptide chains are laterally connected, leading to the formation of bi-directional strands.
This structure is extremely stable.

Classification and nomenclature of the main forms of systemic amyloidosis
By convention, amyloidosis nomenclature specifies the amyloid fibril types by indicating them with two letters: the first letter is always an A (for amyloid) whereas the following letters indicate different chemical types
Simplified classification
1.Primary systemic amyloidosis.
1. No evidence of associated clinical conditions.
2. AL type ("light chain"). Precursor: L chains,that is, the amino-terminal end of the variable region of immunoglobulin light chains (kappa or lambda) as well as the entire chain.
3. Hereditary or familiar amyloidosis. Mutations of plasma proteins including transthyretin (retinol binding protein), apolipoprotein A-I, apolipoprotein A-II, lysozyme, fibrinogen, neutrophil pyrin
· recessive autosomal inheritance: eg. Familial Mediterranean fever (pyrin precursor), familial amyloid polyneuropathy (FAP)
· Dominant autosomal inheritance: eg. constitutive synthesis of mutated forms of transthyretin (TTR)
2. Secondary systemic amyloidosis
1. Reactive. Associated with chronic inflammatory diseases (osteomyelitis, leprosy, tuberculosis, rheumatoid arthritis) and cancer. Precursor: SAA (Serum Acute phase Amyloid, acute phase proteins)
2. Associated to multiple myeloma. Always AL (L chains)
3. Amyloidosis associated with prolonged hemodialysis. AH (Amyloid Hemodialysis). Precursor: MHC beta2-microglobulin.
4. Localized amyloidosis: eg. associated with medullary thyroid carcinoma. AC (Amyloid Calcitonin). Amyloid tumors.
5. Prion-related amyloidosis. PrP (Prion Precursor)
6. Alzheimer's disease-associated amyloidosis. AB (beta-amyloid protein)
AMYLOIDOGENESIS
Rubin's Pathology Copyright 2006
SECONDARY REACTIVE AMYLOIDOSIS
Rubin's Pathology Copyright 2006

SECONDARY IMMUNOCYTIC AMYLOIDOSIS
Rubin's Pathology Copyright 2006
Multiple myeloma-associated amyloidosis (AL amyloidosis)
The figure depicts a normal electrophoretic pattern (left) as well as the electrophoretic pattern of a patient affected by monoclonal gammopathy (right), the latter showing a high and narrow peak in the γ area: the monoclonal component.
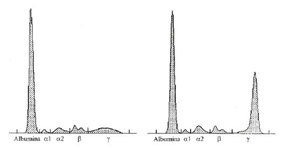
In normal circumstances, the synthesis of heavy and light chains occurs in an orderly manner and in appropriate amounts. The cell assembles heavy and light chains in order to form intact immunoglobulin molecules, leaving no detectable residues behind. In some plasma cell diseases such as myeloma, however, light chain synthesis may greatly exceed heavy chain synthesis. In this case, large amounts of free light chains accumulate in the plasma. Moreover, as light chains are small molecules, they may pass through the glomerular filter of the kidney and are excreted in the urine, where large amounts of these proteins are detected (measured in mg/L). Free light chains in the urine constitute the so-called Bence-Jones proteinuria.
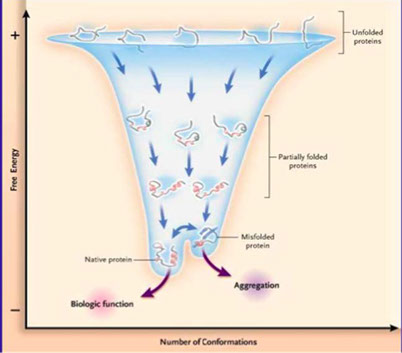
Protein folding
Anatomical and functional alterations of systemic amyloidosis
· The initial phases are characterized by focal perivascular deposits as well as by basement membrane damage.
· The deposition process subsequently extends to the entire interstitial area
· Kidney, liver, spleen and myocardium swelling.
· Kidney damage: chronic renal failure, nephrotic syndrome.
· Cardiac damage: heart failure, arrhythmias
· Liver and spleen damage.
· Presence of amyloid precursors in blood and in urine.
DISTRIBUTION AND ANATOMICAL-PATHOLOGICAL FEATURES
· Heart: "tiger stripes". The most severe manifestation of primary amyloidosis and multiple myeloma. Focal or diffused deposits observed around atrial and ventricular muscle fibers. Very frequent among the elderly. Severe heart failure.
· Kidney: primary glomerular involvement, but peritubular and vascular involvement may also occur. Nephrotic syndrome. Severe functional alterations. Often associated with multiple myeloma.
· Liver: swollen, pale, hardened. Parenchymal atrophy. Distribution between sinusoids and hepatocytes. Very mild functional alterations.
· Spleen: enlarged and hardened, nodular. Focal distribution (around arterioles of the white pulp) or as diffused layers.
· Digestive tract: perivascular, focal or diffused deposits, affecting all districts (including pancreas and gallbladder)
· Eye: cornea and vitreous humor.
· Skin: variably distributed waxy papular lesions
· Nervous system: involvement of peripheral nerves as well as of ganglia, neurofibrillary tangles and vessels (congophilic angiopathy). Senile plaques. Alzheimer's disease. Prion diseases (Kuru, Scrapie, Kreutzfeld-Jacob)
<
>



