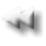Le Gammapatie Monoclonali
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
LE GAMMAPATIE MONOCLONALI
DEFINIZIONE DELLE GAMMAPATIE MONOCLONALI
· Le gammapatie monoclonali sono quadri clinico-laboratoristici caratterizzati dalla proliferazione e accumulo nel midollo osseo di un clone di linfociti B e plasmacellule sintetizzanti immunoglobuline (Ig) identiche per caratteristiche isotipiche (stessa classe di Ig) e idiotipiche (stesso sito di legame con l’antigene nella regione variabile), complete o incomplete, rilevabili nel siero e/o nelle urine.
· Tali Ig prendono il nome di Componente monoclonale (CM)
CLASSIFICAZIONE DELLE GAMMAPATIE MONOCLONALI
GAMMAPATIE MONOCLONALI NEOPLASTICHE
· Mieloma multiplo
· Plasmacitoma localizzato
· Leucemia plasmacellulare
· Macroglobulinemia di Waldenstrom
GAMMAPATIA MONOCLONALE DI SIGNIFICATO NON DETERMINATO (MGUS)
IL MIELOMA MULTIPLO
· 1% di tutte le neoplasie e 10% delle neoplasie ematologiche nei bianchi
· L’ incidenza aumenta con l’età e raggiunge un picco durante la settima decade di vita
· Predominanza sesso M
· Può essere associato ad esposizione a tossici:
- radiazioni ionizzanti
- pesticidi
- benzene
- altri fattori chimici
· Incrementato rischio di MM in soggetti con MGUS (16% a 10 anni)
EVENTO PATOGENETICO
QUADRI CLINICI DEL MM
· Nel 30% dei casi circa la diagnosi di MM è occasionale, sulla base di esami di laboratorio di routine
· Nel restante 70% dei casi è presente una clinica
· I quadri clinici più frequenti sono:
- Patologia scheletrica: da produzione di citochine
- Insufficienza renale: da aumentata escrezione di catene leggere libere urinarie (proteinuria di Bence Jones)
- Morbilità infettiva: secondaria a immunodepressione
PATOLOGIA SCHELETRICA
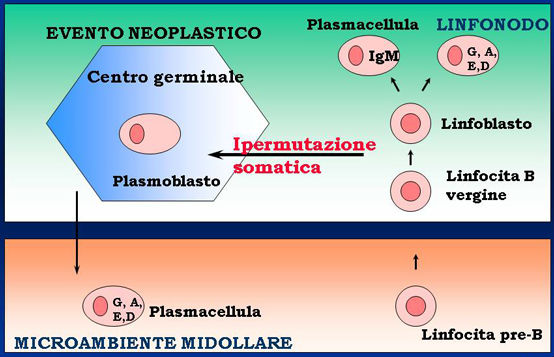
· Presente in circa 70% pazienti alla diagnosi, nella quasi totalità in ricaduta
· Può interessare tutte le ossa, per lo più colonna vertebrale e ossa piatte, sedi di mielopoiesi
· Lesioni osteolitiche, osteoporosi e quadri misti
· Condiziona fortemente la qualità di vita dei pazienti (dolore, rischio di complicanze quali fratture patologiche, crolli vertebrali, compressione midollare)
· Possibilità di sviluppo extra-osseo
Patogenesi della malattia ossea nel MM
Le lesioni osteolitiche del MM sono causate da
· Aumentato riassorbimento osseo per aumentata attività osteoclastica
· Ridotta neoformazione ossea per ridotta attività osteoblastica
Rimodellamento osseo

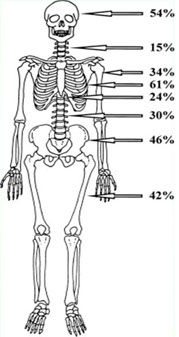
RX SCHELETRO
· Alterato nel 60-70% dei pazienti circa
· Osteolisi circoscritte senza sclerosi periostale, osteopenia, quadri misti
· Evidenza delle lesioni solo quando la massa ossea è ridotta di almeno il 50%
· Possibile sottostima delle lesioni per incapacità di identificare lesioni piccole e basso potere risolutivo nella colonna vertebrale
Pattern
osteoporotico
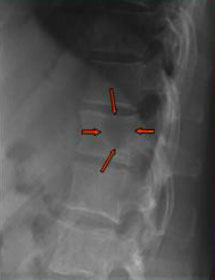
Pattern
osteolitico
RISONANZA MAGNETICA NUCLEARE
· Possibilità di valutazione qualitativa del midollo osseo: lesioni tipicamente ipointense in T1, iperintense in T2, fortemente captanti il mdc (gadolinio)
· Patterns di alterazione: Focale, variegato, diffuso
· Alto potere risolutivo nella colonna vertebrale, dove distingue malattia/osteoporosi e identifica le eventuali complicanze neurologiche
· Valutazione di masse extraossee
· Campo di vista limitato
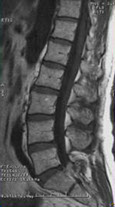
Pattern
normale
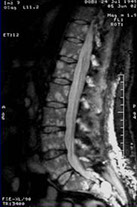
Pattern
focale
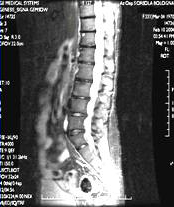
Pattern
diffuso
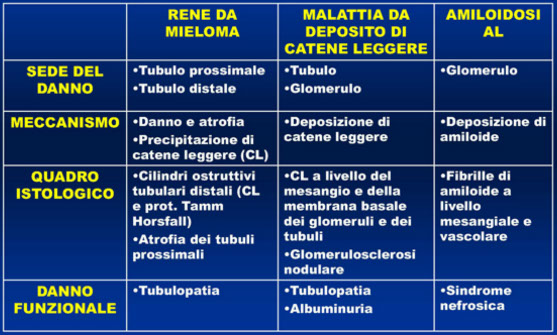
INSUFFICIENZA RENALE
· E’ presente in circa il 50% dei pazienti
· Nel 10-20% dei casi è sintomo d’esordio, nei restanti compare in fase di progressione
· Patogenesi multifattoriale:
- Proteinuria di Bence Jones
- Ipercalcemia
- Sostanze nefrotossiche: FANS, antibiotici, mezzi di contrasto iodati
- Infezioni/disidratazione
· Diversi quadri isto-patologici
INSUFFICIENZA RENALE
Quadri istopatologici e funzionali
SINDROME DA IPERCALCEMIA
· E’ rilevata in circa il 30-40% dei pazienti con MM, nella metà di questi circa all’esordio, in un’altra metà durante il decorso della malattia
· Riconducibile patogeneticamente all’aumentato riassorbimento osseo, con rilascio di calcio nel sangue
· Manifestazioni cliniche: poliuria, polidipsia, anoressia, nausea, vomito, astenia. Se il quadro persiste: disidratazione, confusione, delirio fino al coma (encefalopatia ipercalcemica)
MORBILITA’ INFETTIVA
· Deficit di immunità umorale
· Aumentata incidenza di episodi infettivi (per lo più batterici)
· Poco frequente all’esordio di malattia, progressivamente più rilevante durante il decorso clinico (ricaduta di malattia e immunodepressione post terapia)
Le infezioni sono la principale causa di morte nel paziente con MM
Insufficienza midollare: anemia “secondaria” da cause multiple
· Invasione midollare
· Deficit di eritropoietina
- IRC
- Inadeguata produzione
· Mielosoppressione post terapia
· Produzione di citochine infiammatorie
APPROCCIO AL PAZIENTE CON SOSPETTO MIELOMA
INDAGINI DI LABORATORIO (I)
· Protidemia totale + elettroforesi (iper o ipogammaglobulinemia)
· Dosaggio Ig
· Proteinuria delle 24 ore con dosaggio quantitativo della escrezione delle catene leggere libere monoclonali
· Caratterizzazione della CM (Immunofissazione sierica e urinaria)
· Dosaggio delle catene leggere libere nel siero
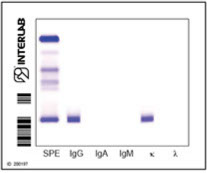
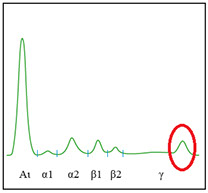
APPROCCIO AL PAZIENTE CON SOSPETTO MIELOMA
INDAGINI DI LABORATORIO (II)
· Aspirato midollare
· Biopsia ossea
· Analisi delle alterazioni cariotipico-molecolari delle plasmacellule
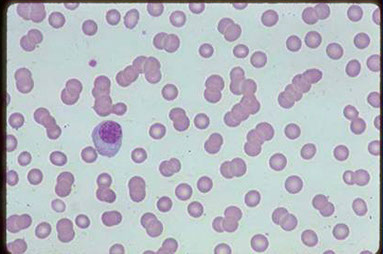
Aspirato midollare di MM
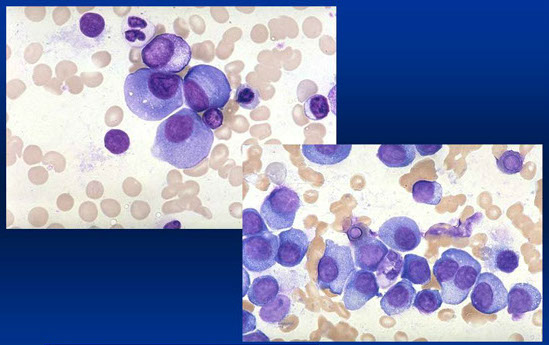

APPROCCIO AL PAZIENTE CON SOSPETTO MIELOMA
INDAGINI STRUMENTALI: METODICHE DI IMAGING
· Rx scheletro in toto (cranio, omeri, emicostati, rachide, bacino, femori)
· RMN
· TAC, 18F-FDG-PET/TC, Scintigrafia con 99mTecnezio Sestamibi
Fondamentali per una corretta stadiazione


FATTORI PROGNOSTICI
ALTERAZIONI CITOGENETICHE
· Monosomia-delezione del cromosoma 13
· Traslocazioni coinvolgenti il gene per la catena pesante delle Ig al locus 14q32 (in particolare t(4;14) e (6;14))
· Delezione del cromosoma 17 (anti-oncogene P53)
· Amplificazioni del gene CKS1B sul cromosoma 1
STRATEGIE TERAPEUTICHE
Quando iniziare la terapia?
· La terapia va iniziata in presenza di MM sintomatico, cioè in presenza di danno d’organo
CRITERI DI ATTIVITA’ DI MALATTIA MM (CRAB)
· C : IPERCALCEMIA (Ca++ ≥ 11.5 gr/dl)
· R : INSUFFICIENZA RENALE (CREA > 1,73 mmol/L)
· A : ANEMIA (Hb più di 2 gr. sotto v.n. o <10 gr/dl)
· B : LESIONI OSSEE (lesioni litiche, osteopenia, fratture patologiche)
STRATEGIE TERAPEUTICHE
· La terapia è modellata in base all’età
· Pazienti anziani > 65 anni: chemioterapia a dosi convenzionali con l’aggiunta dei nuovi farmaci diretti contro il microambiente midollare
· Pazienti giovani< 65 anni: chemioterapia ad alte dosi con trapianto di progenitori emopoietici (autologhi o allogenici) ed eventuale aggiunta dei nuovi farmaci diretti contro il microambiente midollare
LA GAMMAPATIA MONOCLONALE DI SIGNIFICATO NON DETERMINATO (MGUS)
· Diagnosi occasionale in corso di accertamenti laboratoristici
· La clinica è per definizione assente (nessun sintomo o danno d’organo)
· L’incidenza aumenta con l’età
· Non necessita di terapia
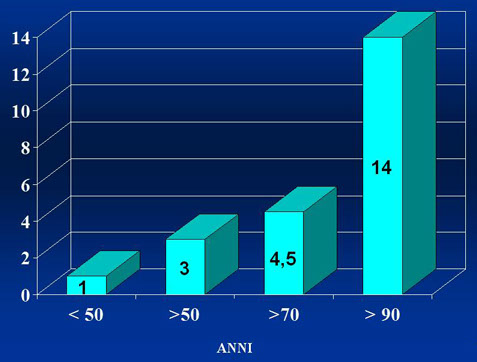
INCIDENZA DELLA MGUS SECONDO L’ETA’
CRITERI DIAGNOSTICI DELLA MGUS
· Concentrazione della componente monoclonale ≤30 g/dL
· Infiltrazione plasmacellulare midollare ≤10%
· Normale concentrazione delle restanti classi immunoglobuliniche
DIAGNOSI DIFFERENZIALE:
MGUS/MM I STADIO/MM SINTOMATICO

PROBABILITA’ DI PROGRESSIONE TRA 1384 PAZIENTI CON MGUS
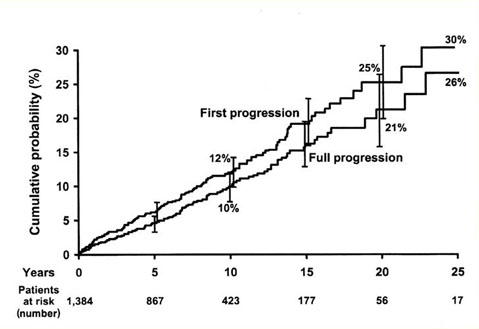
Kyle RA, NEJM 2002
MALATTIA DI WALDENSTRÖM
MALATTIA NEOPLASTICA MONOCLONALE DEL SISTEMA LINFOIDE B CARATTERIZZATA
DA PROLIFERAZIONE ED ACCUMULO DI ELEMENTI LINFOPLASMOCITOIDI COMMISSIONATI ALLA SECREZIONE DI Ig STRUTTURALMENTE OMOGENEE, DI TIPO IgM
MACROGLOBULINEMIA DI WALDENSTROM
· Nel midollo osseo è presente infiltrazione di linfociti maturi, linfociti plasmacitoidi e plasmacellule
· Patologia dell’adulto-anziano (età mediana: 63 anni)
· La diagnosi può essere casuale per occasionale riscontro di incremento delle IgM
· I quadri clinici sono caratterizzati da:
- Sindrome da iperviscosità
- Anemia o citopenie: per infiltrazione midollare
- Linfoadenomegalie-epatosplenomegalia
- Polineuropatia periferica: per attività anticorpale anti-mielina della componente M
EPIDEMIOLOGIA
MALATTIA RARA CHE COLPISCE PREFERIBILMENTE L’ADULTO ANZIANO, SENZA DISTINZIONE DI SESSO O RAZZA




Sindrome da iperviscosità
· Insorge in genere quando IgM> 3 g/dL
· Disturbi visivi (riduzione del visus, emorragie retiniche)
· Disturbi neurologici (cefalea, ronzii auricolari, turbe della concentrazione e della memoria, sonnolenza fino al coma)
· Diatesi emorragica per piastrinopenia e deficit dei fattori della coagulazione
· Trattamento di elezione: plasmaferesi
Manifestazioni neurologiche
· Polineuropatia periferica, prevalentemente sensitiva, a decorso progressivo
· Associata per lo più a IgA o IgM
· 50% casi presente attività anticorpale anti MAG
· Può essere associata a MGUS
· Può essere peggiorata dai farmaci impiegati
Decorso e prognosi
· sopravvivenza mediana: 5 anni
· mancata risposta clinica al trattamento: fattore prognostico sfavorevole
<
>