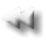Amiloidosi o Beta-Fibrillosi
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
AMILOIDOSI o BETA-FIBRILLOSI
AMILOIDOSI o BETA-FIBRILLOSI
· Deposizione in sede extracellulare di una peculiare sostanza ialina, eosinofila, Lugol e PAS positiva, denominata amiloide da Wirchow
· Colorazione specifica con rosso congo: rossa con birifrangenza verde a luce polarizzata
· Consistenza gommosa, aspetto lardaceo, localizzazione inizialmente perivascolare
· Scarsamente antigenica
· Il deposito può essere localizzato o sistemico e causa atrofia dei parenchimi e disfunzione degli organi interessati da questo processo.
· Sono stati classificati più di 20 tipi di amiloidosi sulla base delle diverse proteine che formano le fibrille di amiloide
· Le forme sistemiche interessano soprattutto rene-miocardio-fegato-milza con aumento delle dimensioni dell’organo
· Costituzione: proteine di composizione amminoacidica eterogenea di struttura a piano beta-pieghettato (diffrazione a raggi X) formanti fibrille, associate a mucopolisaccaridi acidi
Composizione amiloide
1. Componente proteica fibrillare
· Conformazione beta fibrillare costante: due catene polipeptidiche avvolte a spirale e organizzate a foglietto ripiegato
· Composizione biochimica variabile
· Deriva spesso da precursori proteici aventi un ruolo fisiologico
· Scarsamente immunogenica
· Resistente alle proteasi
2. Componente proteica globulare plasmatica
· Glicoproteina globulare (amiloide P sierica) derivante dalla proteina C reattiva
3. Glucosaminoglicani
· Hanno funzione di legame tra le componenti proteiche
· Frequente l’eparansolfato tipico della membrana basale
Arteria da paziente con Amiloidosi colorata con Rosso Congo
Analizzata con A - luce normale
B - luce polarizzata
Rubin Patologia Copyringht 2006 Casa Editrice Ambrosiana
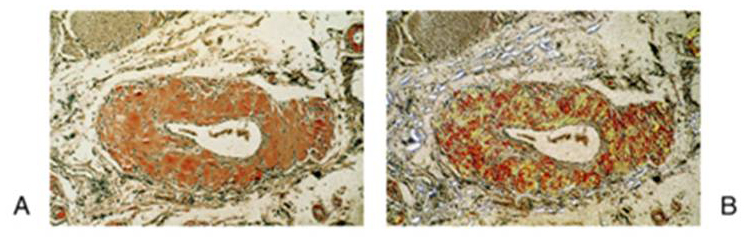
Microscopia ottica
Aspetto ialino di un deposito di amiloide colorata con rosso Congo
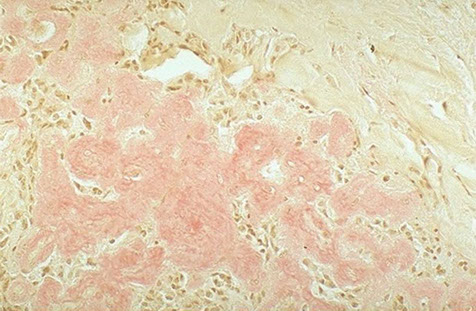
ACCUMULO DI AMILOIDE AL MICROSCOPIO ELETTRONICO
Rubin Patologia - Copyringht 2006 Casa Editrice Ambrosiana
Diffrazione a raggi X
la struttura β-pieghettata delle fibrille di amiloide: le catene polipeptidiche sono legate trasversalmente e formano un nastro disponendosi avanti e indietro
Tale struttura è estremamente stabile

Classificazione e nomenclatura delle principali fiorme di Amiloidosi sistemiche
Per la convenzione, la nomenclatura della amiloidosi prevede che i tipi di fibrilla amiloide siano indicati da due lettere, la prima è sempre una A (amiloide) seguita da una lettera per il diverso tipo chimico
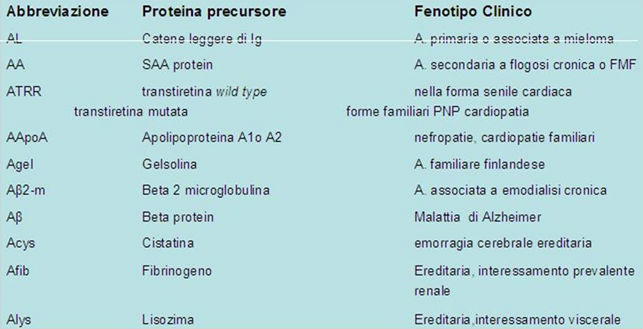
Classificazione semplificata
1. Amiloidosi primarie sistemiche.
1. Nessuna evidenza di malattie associate.
2. Tipo AL (“light chain”). Precursore: Catene L, ossia estremità aminoterminale della regione variabile delle catene leggere delle immunoglobuline (kappa o lambda) o tutta la catena.
3. Amiloidosi eredo-familiari. Mutazioni di protene plasmatiche fra cui transtiretina (proteina legante il retinolo), apolipoproteina A-I, apolipoproteina A-II, lisozima, fibrinogeno, pirina dei neutrofili
· a eredità autosomica recessiva: es. Febbre familiare mediterranea (precursore pirina), polineuropatia amiloide familiare (FAP)
· eredità autosomica dominante: es. sintesi costitutiva di forme mutate di transtiretina (TTR)
2. Amiloidosi secondarie sistemiche
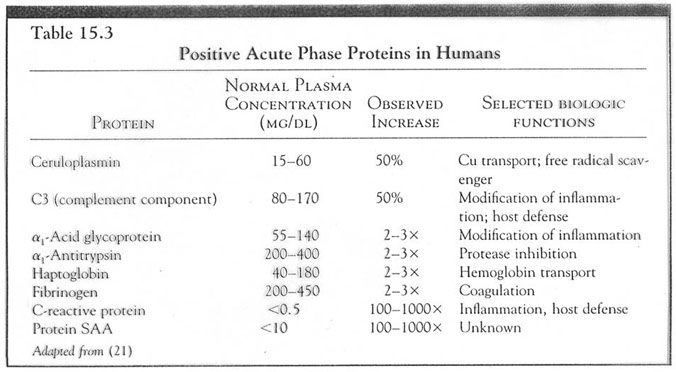
1. Reattive. Associate a malattie infiammatorie croniche (osteomielite, lebbra, TBC, artrite reumatoide) e neoplasie. Precursore: SAA (Serum Acute phase Amyloid, proteine di fase acuta)
2. Associate al mieloma multiplo. Sempre di tipo AL (Catene L)
3. Amiloidosi associata a emodialisi protratta. Tipo AH (Amyloid Hemodialisis). Precursore la beta2 microglobulina del MHC.
4. Amiloidosi localizzate: es. amiloidosi associata al carcinoma midollare della tiroide. Amiloide AC (Amyloid Calcitonin). Tumori amiloidei.
5. Amiloidosi di origine prionica. Tipo PrP (Prion Precursor)
6. Amilodosi associata al morbo di Alzheimer. Tipo AB (Amiloide Beta-proteina)
AMILOIDOGENESI
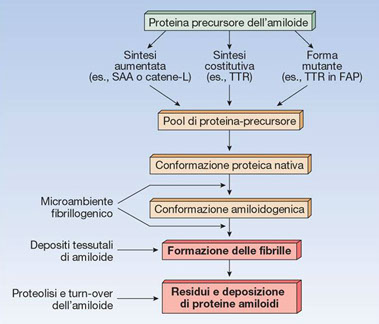
Rubin Patologia Copyringht 2006 Casa Editrice Ambrosiana
AMILOIDOSI SECONDARIE REATTIVE
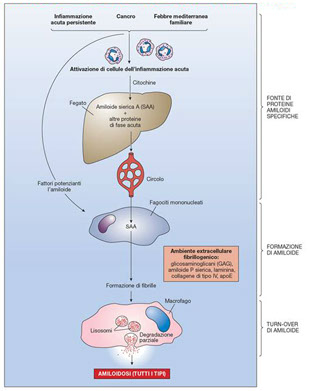
Rubin Patologia Copyringht 2006 Casa Editrice Ambrosiana
AMILOIDOSI SECONDARIE IMMUNOCITICHE
Rubin Patologia Copyringht 2006 Casa Editrice Ambrosiana
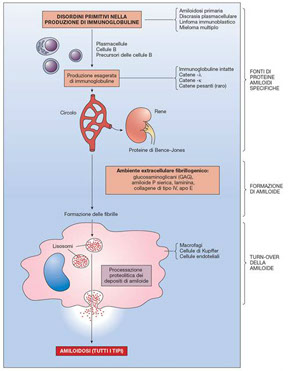
Amiloidosi associata al mieloma multiplo (AL amiloide)
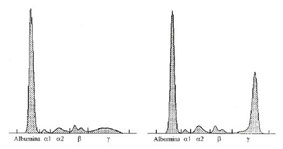
Tracciato elettroforetico normale (a sinistra) e di un paziente con gammopatia monoclonale (a destra). In quest'ultimo si osserva la presenza di un picco alto e stretto in zona γ: la componente monoclonale.
Nell'individuo normale la sintesi di catene pesanti e leggere avviene in modo ordinato e nelle quantità opportune. La cellula assembla catene pesanti e leggere a formare l'intera molecola immunoglobulinica e non vi sono residui apprezzabili. In alcune patologie delle plasmacellule come il mieloma, invece, la sintesi di catene leggere può essere superiore rispetto a quella di catene pesanti. In tal caso grandi quantità di catene leggere libere si accumulano nel plasma. Inoltre, essendo le catene leggere piccole molecole, passano il filtro renale e vengono eliminate nelle urine dove possono essere ritrovate anche in grande quantità (si misurano in mg/litro). Le catene leggere libere urinarie determinano la cosiddetta proteinuria di Bence-Jones.
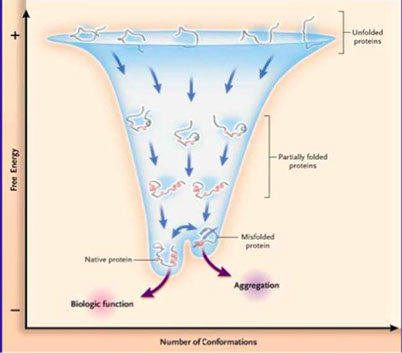
Il ripiegamento delle proteine
Alterazioni anatomo-funzionali delle amiloidosi sistemiche
· Fasi iniziali associate a depositi focali vasali-perivasali con danno alla membrana basale endoteliale.
· Successivamente la deposizione si estende a tutto l’interstizio.
· Ingrossamento di rene, fegato, milza, miocardio.
· Danni renali: insufficienza renale cronica, sindrome nefrosica.
· Danni cardiaci: insufficienza cardiaca, aritmie
· Danni epatici e splenici.
· Presenza dei precursori dell’amiloide nel sangue e nelle urine.
DISTRIBUZIONE E CARATTERISTICHE ANATOMO-PATOLOGICHE
· Cuore: Cuore tigrato. E’ la manifestazione più grave dell’amiloidosi primaria e da mieloma multiplo. Depositi focali o diffusi intorno alle fibre muscolari dell’atrio e del ventricolo. Assai frequente in soggetti anziani. Grave insufficienza cardiaca.
· Rene: interessamento primario dei glomeruli, ma anche peritubulare e vascolare. Sindrome nefrosica. gravi alterazioni funzionali. Spesso associata al mieloma multiplo.
· Fegato: ingrandito, pallido, duro. Atrofia del parenchima. Distribuzione fra i sinusoidi e gli epatociti. Alterazioni funzionali assai lievi.
· Milza: ingrandita e dura, nodulare. Distribuzione focale (nelle arteriole della polpa bianca: a sagù) o diffusa a strati (a prosciutto).
· Tratto digerente: depositi perivascolari, focali o diffusi, interessano ogni distretto (compresi pancreas e colecisti)
· Occhio: umor vitreo e cornea.
· Cute: papule ceree in varie sedi
· Sistema nervoso: interessamento dei nervi periferici, dei gangli, delle maglie neurofibrillari e dei vasi (angiopatia congofila). Placche senili. Morbo di Alzheimer. Malattie da prioni (Kuru, Scrapie, Morbo di Creutzfeld-Jacob)
<
>